
뉴클레오사이드 역전사효소 억제제(NRTIs : Nucleoside Reverse Transcriptase Inhibitors)와 뉴클레오타이드 역전사효소 억제제(NtRTIs : Nucleotide Reverse Transcriptase Inhibitors)의 발견과 발전은 1980년대 에이즈(AIDS : Acquired Immune Deficiency Syndrome) 대유행이 서부를 강타했을 때부터 시작되었다. 뉴클레오사이드 역전사효소 억제제들은 역전사효소( RT : Reverse Transcriptase)를 억제한다. 역전사효소란 인간면역결핍증 바이러스(HIV - Human immunodeficiency Virus)의 유전물질을 복제시키는 효소이다. 첫번째로 개발된 뉴클레오사이드 역전사효소 억제제는 지도부딘(Zidovudine)으로서, 1987년에 미국식품의약국 (FDA : U.S. Food and Drug Administration)의 승인을 받아서 HIV 치료의 첫 디딤돌이 되어주었다. 그 뒤로, 여섯가지의 뉴클레오사이드 역전사효소 억제제와 한가지의 뉴클레오타이드 역전사효소 억제제가 뒷따랐다. 그 둘은 내인성 2'-디옥시 뉴클레오사이드와 뉴클레오타이드( 2'- deoxy-nucleoside and nucleotide)의 유사체들이다. HIV-1 바이러스가 항-HIV 의약품에 지속적으로 노출되면 약물저항성 바이러스들이 생기게 되며, 이는 피할 수 없는 결과이다.
역사
1981년 여름, 최초로 후천면역결핍증후군(에이즈)가 보고되었다. 2년 뒤, 에이즈의 병인학적(etiological) 원인인 인간면역결핍증 바이러스(HIV)가 밝혀졌다.
HIV의 정체가 드러난 후로부터, 효과적인 항레트로바이러스 (antiretroviral) 의약품들이 계속 발전해왔고 HIV에 대한 어마어마한 양의 연구조사가 지속되었다. HIV를 치료하기 위한 항레트로바이러스 의약품은 여섯가지로 분류할 수 있다: 뉴클레오사이드와 뉴클레오타이드 역전사효소 억제제, 비-뉴클레오사이드 역전사효소 억제제, 단백질분해효소 억제제, 진입 억제제, 공동-수용체 억제제(co-receptor inhibitors) 와 통합효소 억제제(integrase inhibitors)이다. 항-HIV 의약품 개발의 주요 토대가 되어준 것은 HIV-1의 역전사 효소이다. 체외에서 항-HIV 효과를 나타낸 최초의 뉴클레오사이드 역전사효소 억제제는 지도부딘이었다. 지도부딘이 1987년에 승인된 이후로, 총 여섯가지의 뉴클레오사이드 그리고 한가지의 뉴클레오타이드 역전사효소 억제제가 미국식품의약국(FDA)으로부터 승인받았다. 미국식품의약국(FDA)으로부터 승인받은 뉴클레오사이드 역전사효소 억제제는 지도부딘, 디다노신(didanosine), 잘시타빈(zalcitabine), 스타부딘(stavudine), 라미부딘(lamivudine), 아바카비르(abacavir) 그리고 엠트리시타빈(emtricitabine)이 있으며, 승인된 뉴클레오타이드 역전사효소 억제제는 테노포비르(tenofovir) (표4 참조) 뿐이다.
HIV-1 역전사효소
기능

대부분의 표준 HIV 의약품 치료요법은 역전사효소를 억제하는 데 초점을 맞춘다. 역전사 효소란, HIV-1을 비롯하여 다른 레트로바이러스들이 자신의 생활사를 완성하기 위해 필요한 효소이다. 역전사 효소는 두가지 주요 기능을 가지고 있다. 첫째로, 그것은 자신의 폴리머레이스(polymerase) 활동으로 바이러스의 유전물질 복제를 제어한다. 그것은 바이러스의 단일-가닥 RNA를 삽입가능한 이중가닥의 DNA로 전환시킨다. 뒤이어, 생성된 DNA는 숙주의 세포내로 이동되어 레트로바이러스의 인터그레이스(integrase)에 의해 게놈(genome)으로 삽입된다. 역전사효소의 다른 역할은 그것의 리보핵산가수분해효소 H(ribonuclease H) 역할이며, 그것은 RNA가 DNA와 헤테로듀플렉스(heteroduplex)를 형성했을 때에만 RNA를 분해한다.
구조
HIV-1 역전사효소는 비대칭의 헤테로다이머(heterodimer)이며, 길이는 1000개의 아미노산으로 되어있고, 두개의 아단위단백질(subunit)로 구성되어있다. 더 큰 아단위단백질은 p66이며 길이는 560개의 아미노산으로 되어있고, 역전사효소의 효소활동을 전부 지니고 있다. 더 작은 아단위단백질은 p51이며 길이는 440개의 아미노산을 가지고 있고, 헤테로다이머를 안정화시키는 것으로 추정된다. 하지만, tRNA 프라이머(primer)가 결합하는데 기여한다는 가능성도 있다. p66 아단위단백질은 두개의 활성자리가 있다: 폴리머레이스와 리보핵산가수분해효소 H이다. 폴리머레이스는 네개의 서브도메인이 있고, 오른손과 유사하여 "손가락(fingers)","엄지(thumb)","이음매(connection)" 그리고 "손바닥(palm)"으로 명명되었다.
활동 메커니즘
뉴클레오사이드와 뉴클레오타이드 역전사효소 억제제들의 활성화는 주로 수동확산(passive diffusion)이나 담체수송(carrier-mediated transport)에 의한 세포내 출입에 의존한다. 뉴클레오사이드 역전사 효소 억제제는 크게 친수성이고 제한적인 세포막 투과성을 가지고 있기 때문에 이 단계는 매우 중요하다. 뉴클레오사이드 역전사 효소 억제제들은 내인성 2'-디옥시 뉴클레오사이드와 뉴클레오타이드 유사체이다. 그들은 모체 원형에서는 비활성화 상태이기 때문에 잇다른 인산화과정이 필요하다.
뉴클레오사이드들은 인산화가 세 번 일어나야 하지만, 뉴클레오타이드는 이미 한 개의 인산기를 보유하고 있기 때문에 두 번의 인산화가 필요하다. 이러한 단계적 활성화 과정은 세포 내에서 일어나며, 여러 가지 효소의 조직화된 연쇄 반응으로 인해 가능해진다. 뉴클레오사이드 유사체의 첫번째 인산화 과정은 보통 속도결정 단계이며, 주로 디옥시뉴클레오사이드 키네이스(kinase)에 의해 촉매화된다. 일인산 뉴클레오사이드 유사체에 두 번째 인산기를 부착하는 과정은 주로 일인산 뉴클레오사이드 인산화효소(NMP kinase)에 의해 촉매된다. 뉴클레오사이드 역전사 효소 억제제의 마지막 인산화 과정은 여러가지의 효소에 의해 촉매화될 수 있다. 예를 들어, 이인산 뉴클레오사이드 인산화효소(NDP kinase)포스포글리세르산 인산화효소(phosphoglycerate kinase), 피루브산 키네이스(pyruvate kinase), 크레아틴 키네이스(creatine kinase)들이 있다. 그들로 인해 뉴클레오사이드 역전사효소 억제제는 저마다의 효과적인 항-바이러스 삼인산 유사체로 변한다. 저마다의 삼인산 형태로, 뉴클레오사이드 역전사효소 억제제들과 한 가지뿐인 뉴클레오타이드 역전사효소 억제제는 저마다 상응하는 디옥시뉴클레오타이드 삼인산과 경쟁하여 발생기의 DNA 사슬에 도입되기 위해 겨룬다.(그림1 참조) 디옥시뉴클레오타이드 기질과는 다르게, 뉴클레오사이드 역전사효소 억제제는 디옥시리보스에 3'-수산기가 없다. 3'-수산기는 잇다른 핵산과 인산다이에스터 결합(phosphodiester bond)을 5'-에서 3'- 방향으로 구성하는 데에 필요하기 때문에, 뉴클레오사이드 역전사 효소 억제제가 일단 DNA에 도입되면 DNA는 더 이상 역전사 효소에 의해 신장될 수 없다. 즉, 종결자(terminator)로서 기능한다.
발견과 발전
HIV치료를 향한 첫 걸음 - 지도부딘
1964년, 미시건 암 재단에서 호르위츠(Horwitz)에 의해 지도부딘(AZT)이 합성되었다. 티민의 디옥시리보오스 고리의 3' 수산화기는 아지도기로 대체되어서 결과적으로 지도부딘이 되었다. 3' 수산화기는 역전사 과정 중 신장하는 DNA에서 다음 뉴클레오타이드에게 부착점을 제공하기 때문에, 수산화기의 부재는 확실한 사슬 종결자로 기능한다. 지도부딘은 티민 대신 사슬에 도입되며 HIV의 유전자 복제를 억제하는 강력한 기능을 가지고 있다. 이 화합물은 1964년에 잠재적인 항암 인자로서 준비되었지만, 효과가 없다고 판명이 났었다. 하지만 1974년, 지도부딘은 레트로바이러스에 대하여 효과가 있다고 보고되었고, 뒤이어 에이즈 대유행이 서부 사회를 1980년대 중반에 강타했을 때 재조명되었다. 하지만, 지도부딘은 상대적으로 독성이 있다. 왜냐하면, 지도부딘은 세포내 효소에 의해 삼인산으로 전환되기 때문에, 감염되지 않은 세포 내에서도 활성화되기 때문이다.
뉴클레오사이드 유사체들의 나아간 발전
다이디옥시뉴클레오사이드(dideoxynucleoside)
| 다이디옥시아데노신 | 다이다노신 | |
|---|---|---|
| 화학적
구조 |

|

|
다이디옥시뉴클레오사이드(dideoxynucleoside)는 당 고리가 2'-수산화기와 3'-수산화기를 모두 가지고 있지 않는 뉴클레오사이드 유사체이다. 지도부딘이 합성된 지 3년 후, 시카고에서 제롬 호르위츠(Jerome Horwitz)와 그들의 동료들은 현재 잘시타빈(Zalcitabine - ddC)이라고 알려진 또 다른 다이디옥시뉴클레오사이드를 개발한다. 잘시타빈은 합성된 피리미딘 뉴클레오사이드 유사체이며 디옥시시티딘과 구조적으로 관련이 있다. 리보스 당의 3'-수산화기는 수소로 대체되어있다. 잘시타빈은 1992년 6월에 HIV-1에 대한 치료효과를 미국식품의약국으로부터 승인받았다.
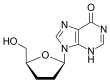
2',3'- 다이디옥시이노신(dideoxyinosine) 혹은 다이다노신(didanosine)은 체내에서 다이디옥시아데노신(dideoxyadenosine)으로 전환된다. 그것의 발전은 매우 긴 역사를 가지고 있다. 1964년에 잘시타빈에 상응하는 아데노신 유사체인 다이디옥시아데노신이 합성되었다. 다이디옥시아데노신은 신장 손상을 초래하기 때문에 다이다노신은 다이디옥시아데노신을 촉매-산성화(enzymatic oxidation)시켜서 생성되었다. ( 표1 참고) 그것은 신장 손상을 초래하지 않으면서도 HIV에 대해 효과적이라고 판단되었다. 다이다노신은 1991년 10월에 HIV-1에 대한 치료효과로 미국식품의약국의 승인을 받았다. 잘시타빈과 다이다노신은 모두 뚜렷한 사슬 종결자로서, 항-HIV 치료에 대해 효과적으로 발전되어왔다. 불행히도, 두 약품 모두 선택성(selectivity)이 낮아서 부작용을 초래하기도 한다.
| 잘시타빈 | 라미부딘 | |
|---|---|---|
| 화학적
구조 |
|

|
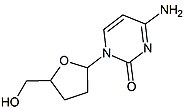
다이디옥시 구조의 나아간 수정은 결국 2',3'-다이디하이드로-3'-디옥시티미딘(2',3'-didehydro-3'-deoxythymidine), 다른말로 스타부딘 (Stavudine, d4T)의 발전을 가지고 왔다. 스타부딘의 활동은 지도부딘과 유사한 것으로 드러났지만, 둘의 인산화 패턴에는 차이가 있다. 지도부딘의 티미딘 키나제(첫번째 인산화에 대한 책임이 있는 효소)에 대한 친화도는 티미딘의 것과 유사하다. 하지만, 스타부딘의 친화도는 700배가량 약하다.
2',3'-다이디옥시-3'-싸이아시티딘( 2',3'- dideoxy-3'-thiacytidine), 다른말로 라미부딘 (Lamivudine, 3TC)은 버나드 벨레우(Bernard Belleau)로부터 발견되었다. 라미부딘의 역사는 1970년대 중반까지 추적되어 올라가는데, 이 때 버나드 벨레우는 당 유도체(derivative)를 면밀히 연구하고 있었다. 라미부딘은 잘시타빈의 황화 유사체로써 개발되었다. (표2 참조) 그것은 초기에 라세믹 혼합물로 합성되었다. ( BCH-189) 분석해보니, BCH-189(2',3'-다이디옥시-3'-싸이아시티딘)의 양과 음의 거울상 이성질체 모두가 체외에서 HIV에 대한 효과가 있다고 드러났다. 라미부딘은 음의 거울상이며 그것은 피리미딘 뉴클레오사이드 유사체이다. 2'-디옥시시티딘의 리보오스 고리의 3' 탄소는 황 원자로 대체되었다. 왜냐하면 그것은 더욱 강한 항-HIV 효과를 나타내며 양의 거울상보다 덜한 독성을 나타냈기 때문이다.
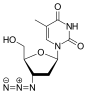
다음으로 뒷따른 것은 2',3'-다이디옥시-5-플루오로-3'-싸이아시티딘(2',3'-dideoxy-5'-fluoro-3'-thiacytidine), 다른말로 엠트리시타빈 (Emtricitabine, FTC)이며, 이것은 라미부딘의 구조적으로 상동(Homologue)이다. 구조적 차이는 라미부딘의 염기 부위에 5-플루오르 수정이다. 그것은 많은 방면에서 라미부딘과 유사하며 HIV-1과 간염 B 바이러스(HBV) 모두에 대해 효과가 있다.
탄소고리 뉴클레오사이드
다이디옥시아데노신의 탄소고리 유사체들은 항-HIV 효과가 있는지 연구되었다. 처음엔 미미한 활동만이 관찰되었다. 많은 뉴클레오사이드 유사체들이 준비되고 관찰되었지만, 오로지 한가지만이 눈에띄는 활동을 하였고 임상적 사용에 적합하도록 충족기준을 만족하였다. 그것이 바로 2' ,3' - 다이디하이드로 유사체인 다이디옥시아데노신이다. 아데닌 고리의 6-아미노기의 질소에 추가된 사이클로프로필기(cyclopropyl)는 화합물의 지질친화도를 높여주었고, 따라서 뇌 침투율을 높여주었다. 결과적인 화합물은 아바카비르(abacavir)라고 알려져있다. (표3 참조) 아바카비르는 1998년 12월에 미국식품의약국으로부터 HIV-1 치료효과를 승인받았다.
이 약물은 체내에서 효과가 있다고 승인받은 항레트로바이러스 중 유일한 구아노신(guanosine) 유사체이다. 그것은 처음엔 아데노신 인산전이효소에 의해 일인산화된다. 그 후, 일인산 화합물은 카보비르 3'-일인산(carbovir 3'-monophosphate)으로 전환된다. 뒤이어, 그것은 완전히 인산화되고 카보비르는 역전사효소에 의해 DNA로 도입되어 사슬 종결자로서 기능한다. 카보비르는 구아노신 유도체이며, 구강상 생물학적 이용 가능성(oral bioavailability)이 매주 낮아 임상적 개발로부터 철회되었다.
| 다이디옥시아데노신 | 다이다노신 | 아바카비르 | |
|---|---|---|---|
| 화학적 구조 |
|
|

|
비고리형 뉴클레오타이드 - 승인된 유일한 뉴클레오타이드 역전사효소 억제제
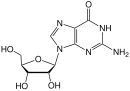
뉴클레오타이드 유사체들은 인산화 과정이 오로지 두 번만 필요하다 (반면에 뉴클레오사이드 유사체들은 세 번이 필요하다.) 인산화과정에서 일어나는 환원은 보다 빠르게 의약품이 전환될 수 있도록 돕는다. 즉, 의약품은 그들의 활성상태 대사물질로 쉽게 전환될수 있다. 이러한 특징을 활용하여 테노포비르와 같은 포스폰산(phosphonate) 뉴클레오타이드 유사체들을 개발하게 되었다. 테노포비르 디소프록실 푸마르산염(Tenofovir DF - Tenofovir Disoproxil Fumarate)는 테노포비르의 프로드러그이다. 테노포비르는 비고리형 아데노신 유도체이다. 화합물의 비고리형 특징과 포스폰산의 존재는 승인된 뉴클레오사이드 역전사효소 억제제들 사이에서 특이한 구조적 특징을 갖게 한다. 테노포비르 디소프록실 푸마르산염은 촉매에 의해 가수분해되어 테노포비르가 되고, 이것이 항-HIV 효과를 나타내게 된다.그것은 2,3-다이하이드록시프로필아데닌(2,3-dihydroxypropyladenine)의 광범위한 항바이러스 활동을 응용하여 개발되고 합성되었다. 테노포비르 디소프록실 푸마르산염은 2001년 10월에 HIV-1에 대한 치료효과로 미국식품의약국의 승인을 받았으며, 최초로 승인받은 뉴클레오타이드 역전사효소 억제제였다.
| 뉴클레오타이드
유사체 |
뉴클레오사이드 유사체 | |||||||
|---|---|---|---|---|---|---|---|---|
 퓨린 유사체 퓨린 유사체
|

피리미딘 유사체 |
퓨린 유사체
|
||||||
| 뉴
클 레 오 사 이 드 |

아데노신 |
 디옥시티미딘 디옥시티미딘
|
 디옥시시티딘 디옥시시티딘
|
아데노신
|
 구아노신 구아노신
|
|||
| 의
약 품 |
 테노포비르 테노포비르
({[(2R)-1-(6-amino-9H- purin-9-yl)propan-2-yl]oxy}methyl) phosphonic acid |
 지도부딘 지도부딘
3´Azido-2´,3´-dideoxythymidine, azidothymidine (AZT) |
 스타부딘 스타부딘
2´,3´-Didehydro- 2´,3´-dideoxythymidine (d4T) |
 엠트리시타빈 엠트리시타빈
(-)-ß-L-3´-thia-2´,3´-dideoxy-5-fluorocytidine ((-)FTC) |
 라미부딘 라미부딘
2´,3´-Dideoxy-3´-thiacytidine (3TC) |
 잘시타빈 잘시타빈
2´,3´-Dideoxycytidine (ddC) |
 디다노신 디다노신
2´,3´-Dideoxyinosine (ddI) |
 아바카비르 아바카비르
(4-(2-amino-6-(cyclopropylamino)- 9H-purin-9yl) cyclopent-2enyl)methanol(ABC) |
why does the table eat the next section heading if nothing is written here?
저항성
현재, HIV-1이 지속적으로 항바이러스 치료제에 노출되면서 약물-저항성 바이러스들이 나타나고 있으며, 이는 피할 수 없는 결과였다. 약물 저항성은 바이러스 감염치료를 할 때 우려되는 임상적인 사안이며, 특히나 HIV를 취급할 때 매우 곤란한 문제이다. 승인된 모든 뉴클레오사이드 역전사효소 억제제들에게서 저항성 돌연변이는 관찰되었다.
뉴클레오사이드 역전사효소 억제제에 대해 약물저항성이 생기는 메커니즘은 두가지가 밝혀져있다: 뉴클레오사이드 역전사효소 억제제가 DNA에 도입될 때 방해받는것, 그리고 도입된 뉴클레오사이드 역전사효소 억제제의 제거이다. 뉴클레오사이드 역전사효소 억제제가 도입될 때 방해받으려면, 사전에 역전사효소의 p66 서브도메인에 돌연변이가 일어나야 한다. 이 돌연변인은 입체 장애를 일으켜서 특정한 약물이 출입하지 못하게 막을 수 있다. 예를 들어, 라미부딘이 역전사과정에 도입되는 것을 막을 수 있다. 도입된 뉴클레오사이드 역전사효소를 제거할 때에는, 저항성 효소들은 기꺼이 억제제를 DNA 사슬의 기질로서 도입한다.뒤이어, 역전사 효소는 중합(polymerization)단계를 이용하여 도입된 뉴클레오사이드 역전사효소 억제제를 제거할 수 있다. 제거과정에서 파이로인산염(pyrophosphate) 제공자가 필요하다. 역전사효소는 뉴클레오사이드 역전사효소억제제의 3'프라이머 말단에 파이로인산염을 결합시켜 그것을 프라이머 DNA로부터 제거시킨다. 환자들에게서 효과적인 HIV-1 복제 억제를 달성하게 위하여, 그리고 약물저항성 바이러스의 등장을 최대한 지연시키기 위해서 약물배합이 사용되고 있다. HAART(Highly Active Antiviral Therapy : 매우 활동적인 항바이러스 치료제)는 뉴클레오사이드 역전사효소 억제제, 뉴클레오타이드 역전사효소 억제제, 비-뉴클레오사이드 역전사효소 억제제, 그리고 단백질분해효소 억제제 등의 항바이러스 약물들을 합쳐서 배합시켰다.
현재 상황
최근에는, 여러 뉴클레오사이드 역전사효소 억제제들이 임상에서부터 전임상(Preclinical)까지 다양한 단계에서 개발중이다. 새로운 뉴클레오사이드 역전사효소 억제제들을 지속하여 연구하는 주요 이유는 독성을 줄이고, 바이러스저항 효율을 증가시키고 항-HIV-1 치료를 간략화시키기 위해서다.
아프리시타빈 (ATC: Apricitabine)
아프리시타빈은 디옥시시티딘 유사체이다. 그것은 라미부딘과 구조적으로 연관이 있으며, 산소의 위치와 황의 위치가 뒤바뀌어있다.다른 뉴클레오사이드 역전사효소 억제제에 비해 체외에서(in vitro) 덜 효과적이지만, 뉴클레오사이드 역전사효소 억제제에 대하여 저항성 돌연변이를 가지는 HIV-1 변종들에 대하여 광범위하게 효과가 유지되기 때문에 유용하다. 아프리시타빈은 뉴클레오사이드 역전사효소 억제제-경험자 환자들을 치료하기 위한 마지막 임상단계에 착수하였다.
엘부시타빈 (L-d4FC: Elvucitabine)
엘부시타빈은 디옥시시티딘 유사체이며, 지도부딘이나 라미부딘과 같은 여러 뉴클레오사이드 유사체들에 저항성을 지니는 HIV에 대해 효과가 있다.그것의 삼인산화 대사물질(triphosphate metabolite)의 농도가 세포내에서 매우 높게 다다를 수 있기 때문이다.엘부시타빈에 대한 임상시험은 현재 보류중이다. 왜냐하면, 몇몇 환자에게서 골수 억제(bone marrow suppression)라는 부작용이 관찰되었기 때문이다. 약물 주입 후, 빠르면 2일 내로 CD4+ 세포의 수치가 떨어지는 것이 관찰되었다.
암독소비르 ( DAPD: Amdoxovir )
암독소비르는 좋은 생물학적 가용능을 가진 구아노신 유사체 - 뉴클레오사이드 역전사효소 억제제 프로드러그이다. 그것은 세포내에서 아데노신 디아미나제(adenosine deaminase)에 의하여 아민기가 제거되어 디옥솔레인 구아닌( DXG: dioxolane guanine)이 된다. 디옥솔레인 구아닌 삼인산(DXG-triphosphate)은 약물의 활성화된 구조로 DAPD-triphosphate보다 효과가 강하다.암독소비르는 현재 임상 2단계에 있다.
Amdoxovir is currently in phasa II clinical trials.
라시비르 (RCV: Racivir )
라시비르는 엠트리시타빈(FTC)의 두가지 β-거울이성질체들을 섞은 라세믹혼합물이며, (-)-FTC 와 (+)-FTC 로 구성되어 있다. 라시비르는 탁월한 구강상의 생물학적 가용능을 가지고 있고, 하루에 오로지 한번만 섭취해도 된다는 이점이 있다. 라사비르는 두개의 뉴클레오사이드 역전사효소 억제제를 혼합할 때 사용되며, 이 때 항바이러스 효과는 보장된 것으로 알려져있다. 라시비르는 현재 임상 2단계에 있다.
| 약물 후보 | 아프리시타빈 | 엘부시타빈 | 암독소비르 | 라시비르 |
|---|---|---|---|---|
| 화학적 구조 |

|

|

|

|
| 개발단계 | 임상개발의 마지막 단계 | 보류 중 | 임상 2 단계 | 임상 2 단계 |
추가적으로 여러가지의 뉴클레오사이드 역전사효소 억제제들이 더 개발중이다. 스폰서들이 IND( Investigational New Drug) 승인요청을 제기하여 미국식품의약국으로부터 승인받았거나, 혹은 약물이 각기 다른 임상단계에 있다. 개발중인 몇개의 뉴클레오사이드 역전사효소 억제제들은 약학적으로 매력적인 특징을 나타내고 있어서 새로운 약물이 필요한 환자들에게 바람직한 치료제가 되어 줄 전망이다.
상호 참조
- 항레트로바이러스 의약품: Antiretroviral drug
- CCR5 수용체 길항제의 발견과 발전: Discovery and development of CCR5 receptor antagonists
- 비뉴클레오사이드 역전사효소 억제제의 발견과 발전: Discovery and Development of Non-Nucleoside Reverse-Transcriptase Inhibitors
- HIV 단백질분해효소 억제자의 발견과 발전: Discovery and Development of HIV Protease Inhibitors
- 역전사효소 억제제: Reverse-transcriptase inhibitor
- 단백질분해효소 억제제: Protease inhibitor
- 진입 억제제: Entry inhibitor
- HIV 단백질분해효소 억제자의 발견과 발전: Discovery and development of HIV protease inhibitors
- CCR5 수용체 길항제의 발견과 발전: Discovery and development of CCR5 receptor antagonists